If you choose to do the library preparation with us, we will quality control the libraries using the Tapestation from Agilent to evaluate the fragment sizes and perform QPCR to accurately measure their concentration. We will pool the libraries in equimolar accordingly. Should you choose to prepare the libraries yourself you have the option to do the library QC yourself or have it done by us.
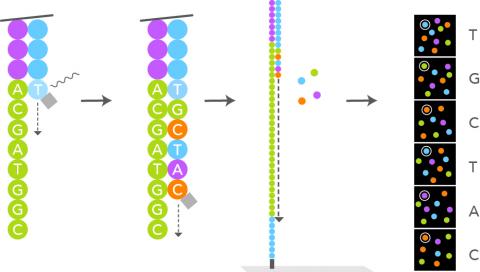
A good successful sequencing run is determined by:
1- The amount of data generated.
The aim is to maximise the sequencing run and reach as much as possible the instrument maximum output capacity. This can be measured by the cluster density of the run.
2- The balancing of the samples.
It is critical to measure with accuracy the individual libraries before and after pooling. This will determine the total output of a run and the even representation of the individual samples so that all have been sequenced equally and have the required coverage or number of reads.
3- The quality of the data.
For a quick overview of the quality of the raw data, the sequencer will generate a control board. The important QC scores to look for are the number of clusters, the number of clusters passing filter and the Q30 which tells you the number of sequences with less than 1 error per 1000 bases sequenced.
In any sequencing project whether it is DNA or RNA, it is one of the critical point to determine during the design phase of the experiment the required genome coverage or the number of reads. The resolution needed will depend on the application and objective and in turn this will determine how much sequencing or number of lanes is required.
We have spent time developing and optimising our methods in the laboratory to get this step spot on in order to provide you with as much data as possible and meet the required depth of sequencing for as little sequencing as possible. This is not a trivial step as there are a number of variable factors including the nature of the libraries, the kit used, inherent pipetting errors particularly as we work with very small volumes and include multiple dilution steps, the inaccurate estimation of the average fragment size. This can lead to under-clustering or unbalanced pooling of the libraries and the need of extra run(s) and expense. Although we do our best to avoid such situations some factors are out of our control and might lead to under of over clustering.